Today we will perform an RNASeq analysis on the effects of dexamethasone (hereafter “dex”), a common steroid, on airway smooth muscle (ASM) cell lines.
Data Import
We need two things for this analysis:
countData: a table with genes as row and samples/experiments as columns
colData: metadata about the columns (i.e. samples) in the main countData object.
id dex celltype geo_id
1 SRR1039508 control N61311 GSM1275862
2 SRR1039509 treated N61311 GSM1275863
3 SRR1039512 control N052611 GSM1275866
4 SRR1039513 treated N052611 GSM1275867
5 SRR1039516 control N080611 GSM1275870
6 SRR1039517 treated N080611 GSM1275871
Check on metadata counts corespondance
We need to check that the metadata matches the samples in our count data.
ncol(counts) ==nrow(metadata)
[1] TRUE
colnames(counts) == metadata$id
[1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
Q1. How many genes are in this dataset?
nrow(counts)
[1] 38694
There are 38,694 genes in this dataset.
Q2. How many “control” samples are in this dataset?
sum(metadata$dex =="control")
[1] 4
There are 4 “control” samples in this dataset.
Analysis Plan…
We have 4 replicates per condition (“control” and “treated”). We want to compare the control vs the treated to see which genes expression levels change when we have the drug present.
“We will go row by row (gene by gene) and see if the average value in control columns is different than the average value in treated columns”
Step 1. Find which columns in counts correspond to “control” samples.
# The indices (i.e. positions) that are "control"control.inds <- metadata$dex =="control"
Step 2. Extract/select these columns
# Extract/select these "control" columns from countscontrol.counts <- counts[,control.inds]
Step 3. Calculate an average value for each gene(ie. each row).
#Calculate the mean for each gene (i.e. row)control.mean <-rowMeans(control.counts)
Q3. How would you make the above code in either approach more robust? Is there a function that could help here?
We can make the code more robust by not hard-coding the number of control samples. A function that could help here could be rowMeans().
Q. Do the same for “treated” samples - find the mean count value per gene.
Q4. Follow the same procedure for the treated samples (i.e. calculate the mean per gene across drug treated samples and assign to a labeled vector called treated.mean)
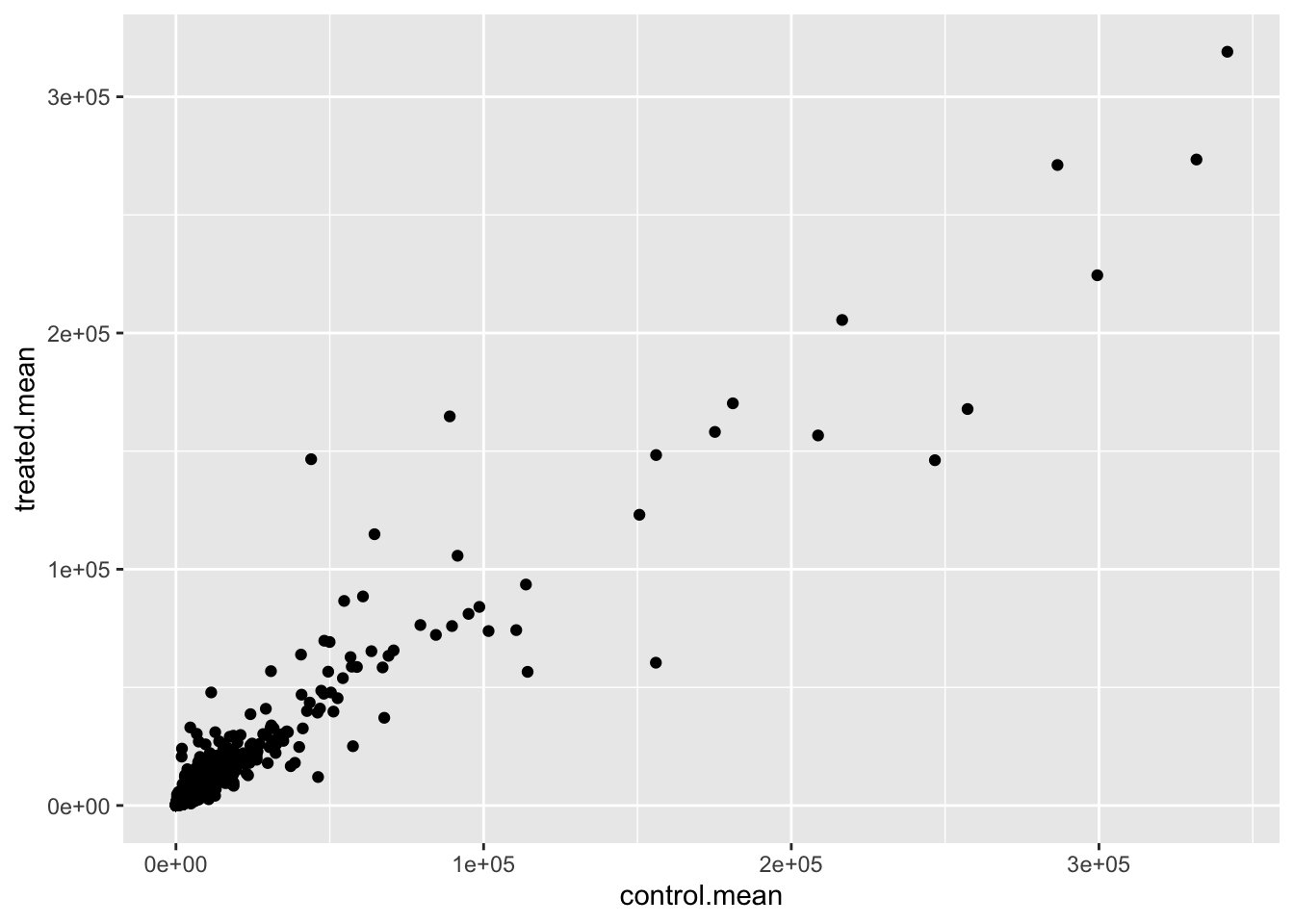
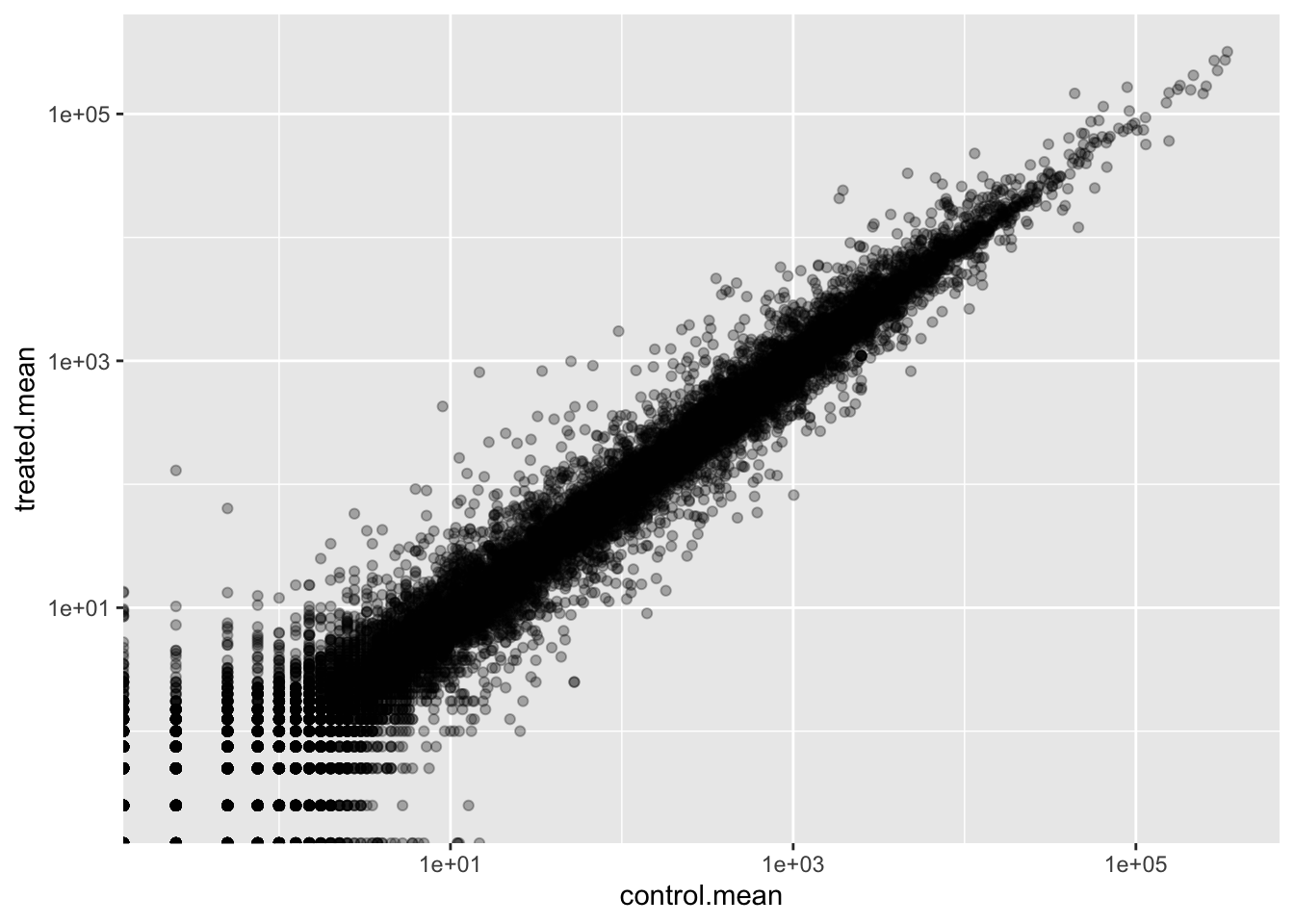
Q5 (a). Create a scatter plot showing the mean of the treated samples against the mean of the control samples. Your plot should look something like the following.
Warning in scale_x_log10(): log-10 transformation introduced infinite values.
Warning in scale_y_log10(): log-10 transformation introduced infinite values.
Log2 units and fold change
If we consider “treated” / “control” counts we will get a number that tells us the change.
# No changelog2(20/20)
[1] 0
# A doubling in the treated vs controllog2(40/20)
[1] 1
# log2(10/20)
[1] -1
Q7. What is the purpose of the arr.ind argument in the which() function call above? Why would we then take the first column of the output and need to call the unique() function?
The purpose of the arr.ind argument is that arr.ind = TRUE makes which() return the row and column indexes. The first column gives the row indeces and unique() makes it so each gene is removed once.
Q. Add a new colum log2fc for log2 fold change of treated/control to our meancounts object.
Q8. Using the up.ind vector above can you determine how many up regulated genes we have at the greater than 2 fc level?
250 up-regulated genes.
Q9. Using the down.ind vector above can you determine how many down regulated genes we have at the greater than 2 fc level?
367 down-regulated genes.
Q10. Do you trust these results? Why or why not?
No, I don’t trust these results because there are many other variables such as normalization and variance/dispersion that are not accounted for.
Remove zero count genes
Typically we would not consider zero count genes - as we have no data about them and they should be excluded from further consideration. These lead to “funky” log2 fold change values (e.g. divide by zero errors etc.)
DESeq analysis
We are missing any measure of significance from the work we have so far. Let’s do this properly with the DESeq2 package.
library(DESeq2)
The DESeq2 package, like many bioconductor packages, wants it’s input in a very specific way - a data structure setup with all the info it needs for the calculation.
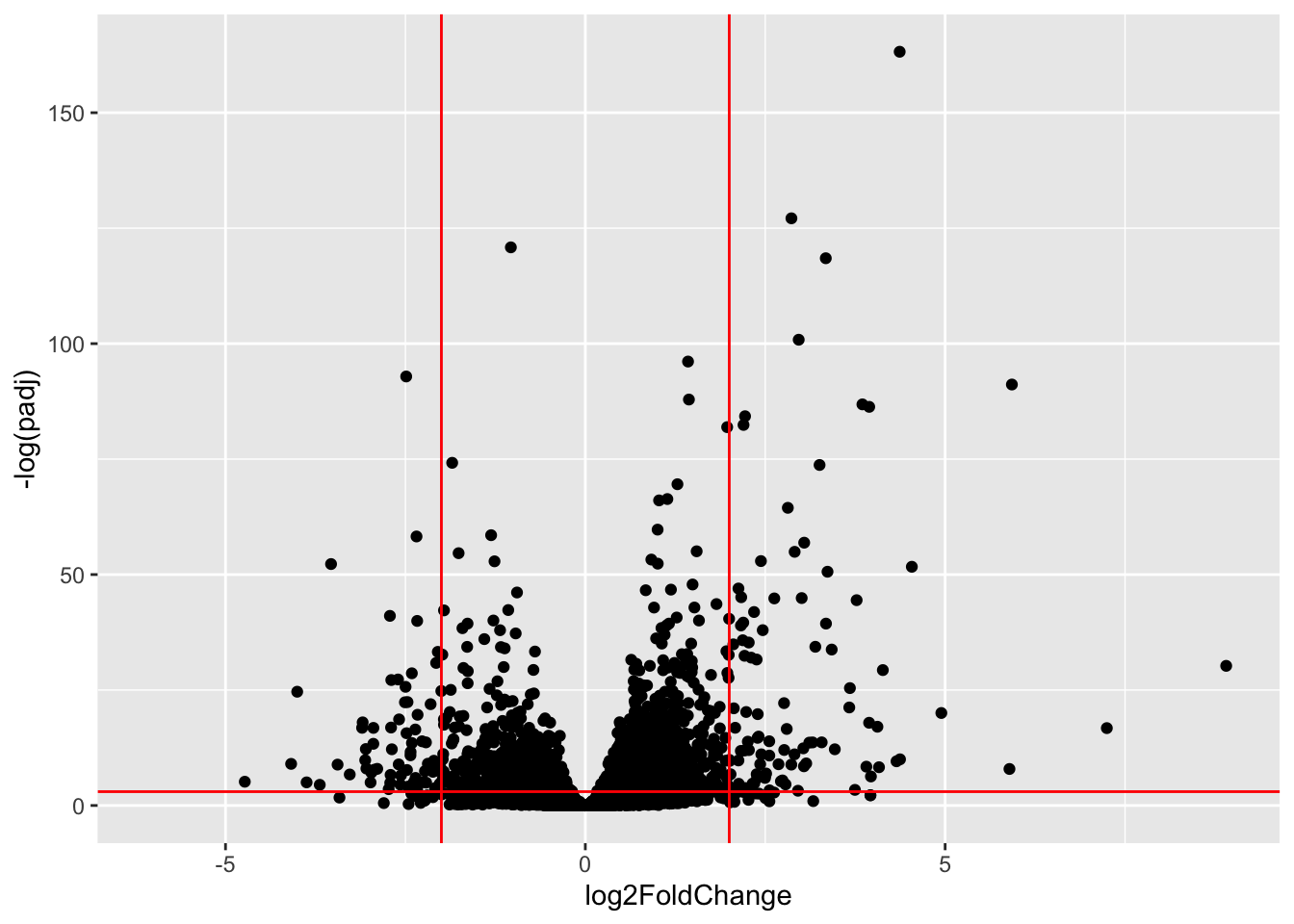
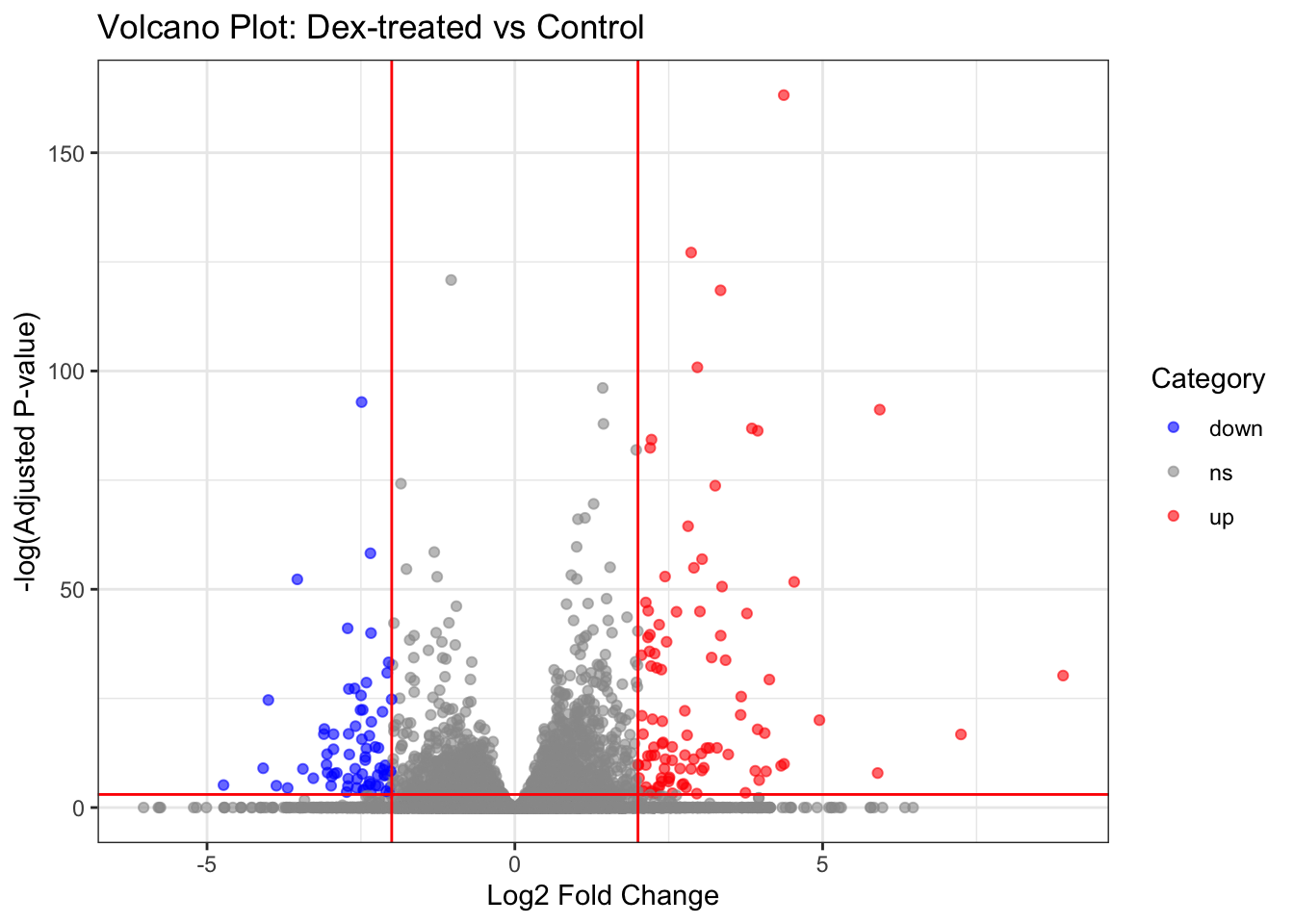
Warning: Removed 13436 rows containing missing values or values outside the scale range
(`geom_point()`).
Save our results to a CSV file
write.csv(res, file="results.csv")
Add Annotation Data
To make sense of our results, we need to know what the differentiated expressed genes are and what biological pathways and processes they are involved in.
Let’s start by mapping our ENSEMBLE ids to the more conventional gene SYMBOL.
head(res)
log2 fold change (MLE): dex treated vs control
Wald test p-value: dex treated vs control
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 747.194195 -0.350703 0.168242 -2.084514 0.0371134
ENSG00000000005 0.000000 NA NA NA NA
ENSG00000000419 520.134160 0.206107 0.101042 2.039828 0.0413675
ENSG00000000457 322.664844 0.024527 0.145134 0.168996 0.8658000
ENSG00000000460 87.682625 -0.147143 0.256995 -0.572550 0.5669497
ENSG00000000938 0.319167 -1.732289 3.493601 -0.495846 0.6200029
padj
<numeric>
ENSG00000000003 0.163017
ENSG00000000005 NA
ENSG00000000419 0.175937
ENSG00000000457 0.961682
ENSG00000000460 0.815805
ENSG00000000938 NA
We will use two bioconductor packages for this “mapping” AnnotationDbi and org.Hs.eg.db.
We will first need to install these from bioconductor with BiocManager::install("")
res$symbol <-mapIds(org.Hs.eg.db,keys =rownames(res), # Our idskeytype ="ENSEMBL", # Their formatcolumn ="SYMBOL") # What I want to translate to
'select()' returned 1:many mapping between keys and columns
head(res)
log2 fold change (MLE): dex treated vs control
Wald test p-value: dex treated vs control
DataFrame with 6 rows and 7 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 747.194195 -0.350703 0.168242 -2.084514 0.0371134
ENSG00000000005 0.000000 NA NA NA NA
ENSG00000000419 520.134160 0.206107 0.101042 2.039828 0.0413675
ENSG00000000457 322.664844 0.024527 0.145134 0.168996 0.8658000
ENSG00000000460 87.682625 -0.147143 0.256995 -0.572550 0.5669497
ENSG00000000938 0.319167 -1.732289 3.493601 -0.495846 0.6200029
padj symbol
<numeric> <character>
ENSG00000000003 0.163017 TSPAN6
ENSG00000000005 NA TNMD
ENSG00000000419 0.175937 DPM1
ENSG00000000457 0.961682 SCYL3
ENSG00000000460 0.815805 FIRRM
ENSG00000000938 NA FGR
Q11. Run the mapIds() function two more times to add the Entrez ID and UniProt accession and GENENAME as new columns called res\(entrez, res\)uniprot and res$genename.
Q. Can you add “GENENAME” and “ENTREZID” as new columns to res as “name” and “entrez”?
res$name <-mapIds(org.Hs.eg.db,keys =rownames(res), # Our idskeytype ="ENSEMBL", # Their formatcolumn ="GENENAME") # What I want to translate to
'select()' returned 1:many mapping between keys and columns
res$entrez<-mapIds(org.Hs.eg.db,keys =rownames(res), # Our idskeytype ="ENSEMBL", # Their formatcolumn ="ENTREZID") # What I want to translate to
'select()' returned 1:many mapping between keys and columns
head(res)
log2 fold change (MLE): dex treated vs control
Wald test p-value: dex treated vs control
DataFrame with 6 rows and 9 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 747.194195 -0.350703 0.168242 -2.084514 0.0371134
ENSG00000000005 0.000000 NA NA NA NA
ENSG00000000419 520.134160 0.206107 0.101042 2.039828 0.0413675
ENSG00000000457 322.664844 0.024527 0.145134 0.168996 0.8658000
ENSG00000000460 87.682625 -0.147143 0.256995 -0.572550 0.5669497
ENSG00000000938 0.319167 -1.732289 3.493601 -0.495846 0.6200029
padj symbol name entrez
<numeric> <character> <character> <character>
ENSG00000000003 0.163017 TSPAN6 tetraspanin 6 7105
ENSG00000000005 NA TNMD tenomodulin 64102
ENSG00000000419 0.175937 DPM1 dolichyl-phosphate m.. 8813
ENSG00000000457 0.961682 SCYL3 SCY1 like pseudokina.. 57147
ENSG00000000460 0.815805 FIRRM FIGNL1 interacting r.. 55732
ENSG00000000938 NA FGR FGR proto-oncogene, .. 2268
write.csv(res, file ="results_annotated.csv")
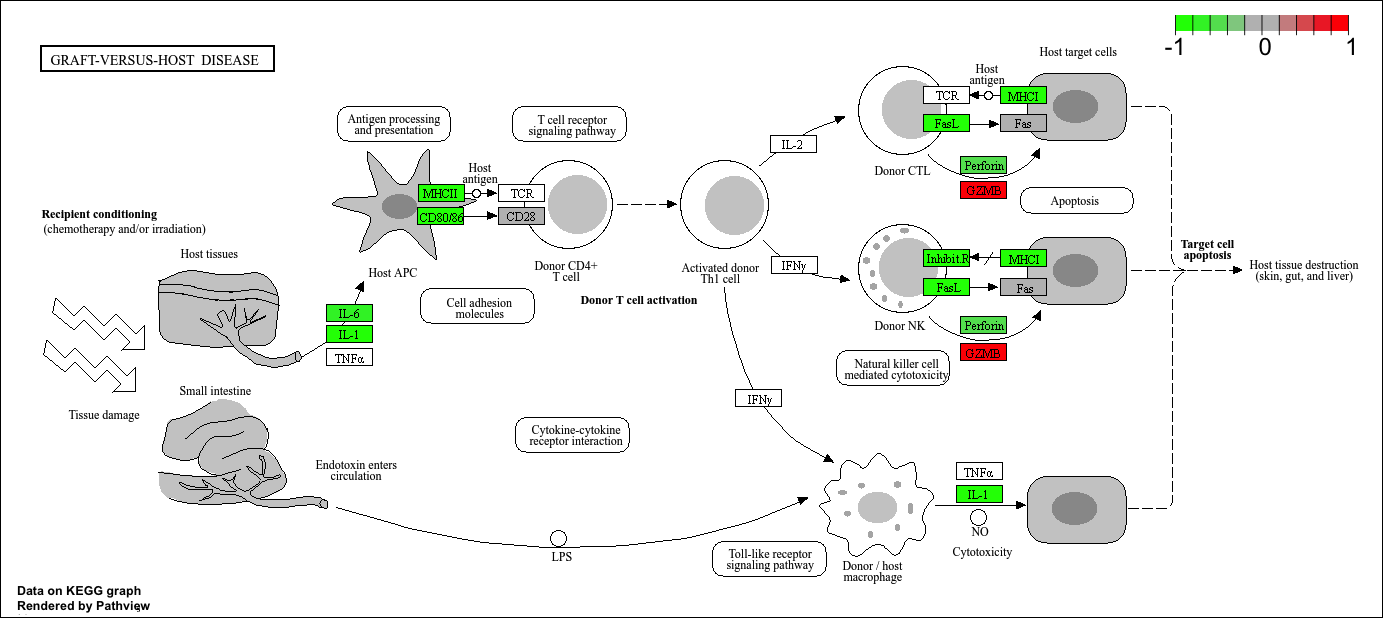
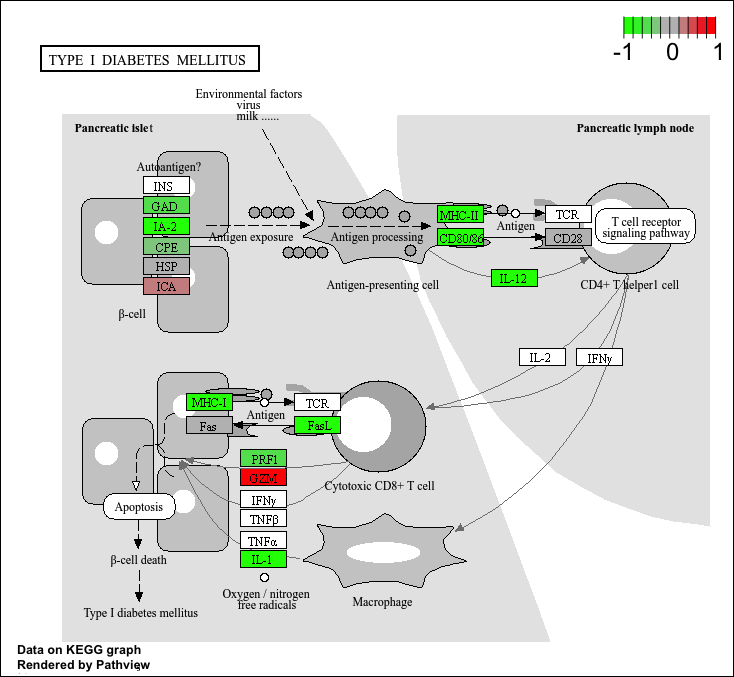
Pathway analysis
Now that we know the gene names (gene symbols) and their entrez IDs, we can find out what pathways they are involved in. This is called “pathway analysis” or “gene set enrichment”.
We will use the gage package and the pathviewer package to do this analysis (but there are loads of others).
To run our pathway analysis we will use the gage() function. It wants to main inputs: a vector of importance (in our case the log2 fold change values); and the gene sets to check overlap for.